作为社会实践的作业之一,讲解一下 AlphaFold3 的一些知识
由于 Alphafold3 是在 Alphafold2 的基础之上增加了新的功能,因此先讲一下 AlphaFold2,再讲 AlphaFold3 的改进
AI for proteins
基于氨基酸序列预测蛋白质三维结构一直是生物界难题,也是 AI4Science 在生化领域的主攻方向之一 这方面预测方法主要分两派:
- 物理办法:模拟原子作用力,虽然最完美但是计算过于困难,模拟的精度很有限,因此进展不多
- 进化办法(主流,AI 预测采用的办法): 通过蛋白质的演变规律来推导结构性质
- 同源建模: 假如有一个氨基酸序列相似且结构已知的蛋白,可以直接以此为模版构建新的蛋白模型
- 共进化分析(AI任务): 通过分析 MSA(这个蛋白质和别的 “进化亲戚” 蛋白质的序列信息在计算机里面对齐形成的一个表格) 来计算残基对的共进化分数(序列上两块氨基酸的规律),从而预测哪些残基对是相互接触的,这个方法叫 接触图预测
关于MSA:AI给的例子
假如这个表是人类血红蛋白和其他动物血红蛋白的氨基酸序列对比形成的MSA
| 物种 | … | 位置 62 | 位置 63 | 位置 64 | … |
|---|---|---|---|---|---|
| 人 | … | A | H | G | … |
| 黑猩猩 | … | A | H | G | … |
| 狗 | … | G | H | G | … |
| 鸡 | … | T | H | G | … |
| 鱼 | … | S | H | A | … |
通过扫描这个巨大的表格,AlphaFold可以获得两种决定性的生物学线索:
线索一:发现功能核心(保守性)
请看表格中的第63列。你会发现,从人类到鱼类,尽管其他位置的氨基酸变来变去,但第63位的氨基酸始终是H(组氨酸)。这个位置在进化中从未改变,我们称之为高度保守。
- 生物学意义:这告诉AlphaFold,这个“H”氨基酸是血红蛋白的“命根子”。事实上,这个组氨酸在三维结构中正扮演着一个至关重要的角色:它像一只手,直接抓着血红蛋白核心的“血红素”(一个含铁的结构),而氧气正是与这个铁结合的。如果这个“H”改变了,整个蛋白质就会失去运输氧气的功能。AlphaFold看到这种绝对的保守性,就能推断出:这个氨基酸一定是功能中心的一部分,其3D位置必须非常精确。
线索二:发现三维空间中的“搭档”(共进化)
现在想象一下,在序列的另一个部分,比如第90位和第110位。AlphaFold可能会发现这样一种模式:
-
在哺乳动物中,第90位是一个体积大的氨基酸,而第110位是一个体积小的。
-
但在鸟类和鱼类中,如果第90位突变成了一个体积小的氨基酸,那么第110位也总是相应地突变成一个体积大的氨基酸,仿佛是为了填补空间。
这种“你变我也变”的关联变化,就叫做共进化。
- 生物学意义:即使第90位和第110位在1D序列上相隔很远,但它们这种协同变化的模式是一个强有力的证据,表明它们在折叠后的三维空间中是物理上相互接触的伙伴。一个变大了,另一个就得变小来腾出空间。
AlphaFold 2的突破
之前 AI 主要是参考 CNN 架构,把 MSA 矩阵当成一个图像,深度学习产出一个 接触图,用这个图的几何约束指导传统蛋白质折叠算法构建 3D 模型。虽然有一定成效,模型并不能直接生成一个 3D 模型
AlphaFold2 开创了第一个可以直接输出原子 3D 坐标的端到端模型,建设了 3D空间推理的新范式,进而催生了例如蛋白质复合物、细胞分子系统(AlphaFold3)的其他空间预测模型
AlphaFold2 pipeline
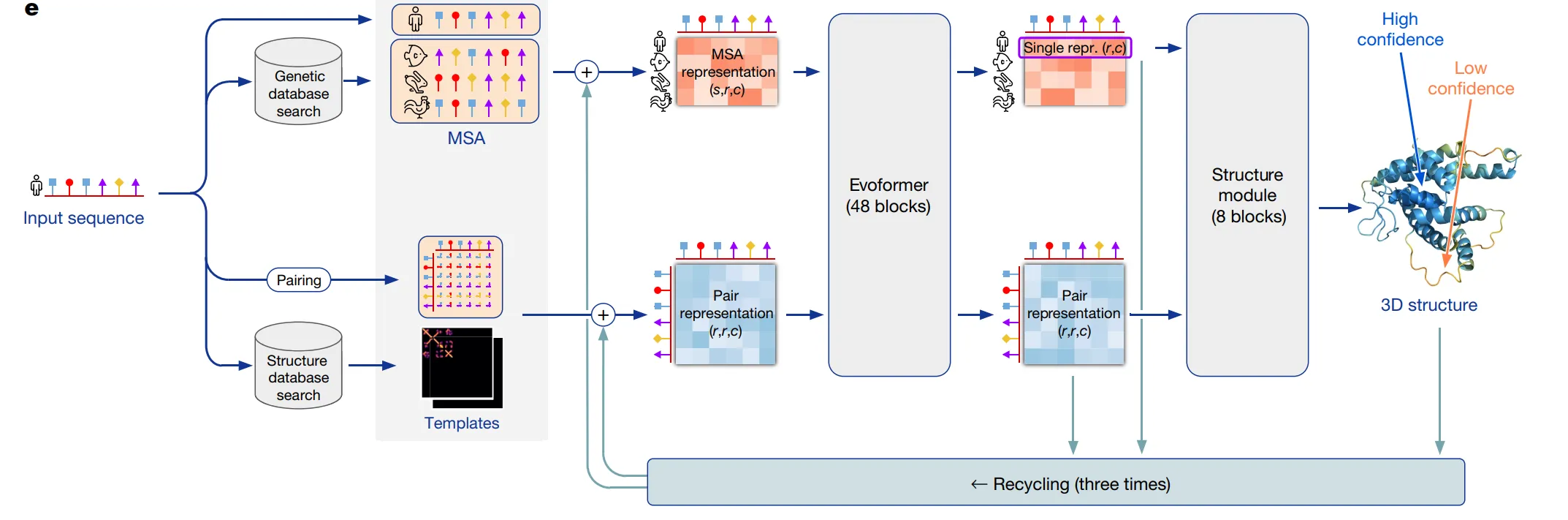
Input
人工输入目标蛋白质的一维的氨基酸序列
例如,输入一串字母 VHLTPEEKSAVTALWGKVNVDEVGGEALGR....
Feature representation
程序自动检索数据库并生成:
- 多序列比对(MSA):程序会自动去基因数据库搜索 与目标蛋白在进化上相关的氨基酸序列集合,生成一个MSA
- 同源蛋白(templates): 搜索蛋白质结构数据库,寻找已知结构的有相似序列的蛋白质作为模版(可选输入)
然后将这些信息表征为 Tensor:
MSA—— 形状为 的张量 M,三个参数分别代表MSA序列数,蛋白质的氨基酸序列长度,以及通道数(可以认为是把每个氨基酸都变成一个长度为 的向量,存储各种氨基酸特征信息,如化学特性)
Pair:可以理解成一个氨基酸序列的关系网,储存序列中任意两个氨基酸之间的空间关系以及各种化学性质 例如假设我们有一个非常短的蛋白质,序列只有5个氨基酸,我们称之为 A-B-C-D-E。那么它的Pair表征就是一张 5x5 的地图:
| A | B | C | D | E | |
|---|---|---|---|---|---|
| A | A-A | A-B | A-C | A-D | A-E |
| B | B-A | B-B | B-C | B-D | B-E |
| C | C-A | C-B | C-C | C-D | C-E |
| D | D-A | D-B | D-C | D-D | D-E |
| E | E-A | E-B | E-C | E-D | E-E |
注意,这个表格的每一个格子都对应一个向量
模型利用原始一维序列+可能的模版,构建tensor , 相关参数含义同上。注意这一步的表征很初级
==Evoformer ==(Main innovation)
接受两个MSA(M),Pair (Z) 作为输入
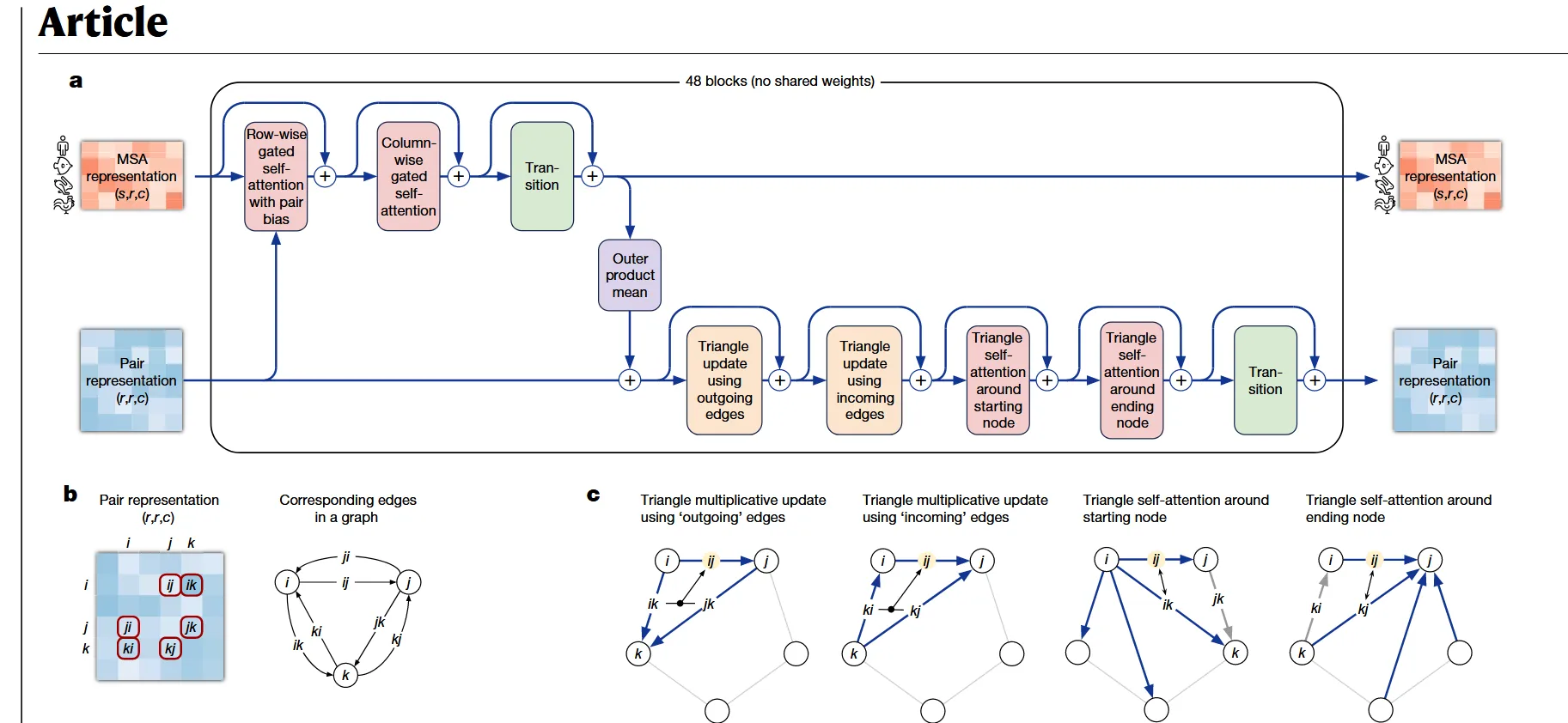
从一维氨基酸序列到三维蛋白质结构,直接通过大量数据进行映射是很困难的,因此这里作者的想法是 利用已有数据构建和推理一个蛋白质中氨基酸(蛋白质残基)之间的“关系网”,也就算一个图结构 图的节点是氨基酸残基,图的边是每一对氨基酸之间的空间关系
而 EvoFormer 的意图,就是不仅让 MSA 和 Pair 内部信息能够充分交换,更能建立互相的信息交换
我们可以把这个架构分成三个阶段:
-
理解MSA: 通过 行向(对一个氨基酸序列中的不同氨基酸关系) 和 列向(对不同氨基酸序列同一位置氨基酸) 的注意力机制,分析氨基酸的进化模式和互相关系
[!一note] 一个创新点 行向注意力计算时会引入来自 Pair 的几何信息作为偏置 可能的算法实现:原先注意力机制计算出 ,但是在偏置之下会将 的 embedded vector 通过 linear projection (乘一个矩阵) 得到一个 , 最终的 logit 是这两个的叠加 比喻:这相当于在说:“嘿,进化空间,当你们在分析序列时,别忘了参考一下我们几何空间当前画的草图。如果草图显示残基A和B离得很近,你们就应该更关注它们之间的联系。” 这实现了用当前的3D假设来指导对1D进化信息的解读。
-
MSA和 Pair 的交流:MSA 给 Pair 传递遗传进化信息,完善 Pair 的表征 算法实现:对MSA的每一个序列,计算每一个序列的 残基 的表征向量的 外积(结果是一个 的矩阵) 这样先得到一个四维的 tensor, 再用 view+线性变换成和 Pair 一样的三维 tensor 再贴上去 不用内积可能是为了更好的计算两个向量的匹配度 这个复制过来的tensor会和原来的偏置tensor进行叠加
-
Pair的三角更新: 可以理解成一个对关系图的审查步骤,之前的关系图都没有引入第三方审查,可能会违背物理规律,例如说A到B的距离是1,B到C距离为1,C到A距离却是10
对于两个残基 , 遍历所有第三个残基 k 来计算更新 i 和 j 之间的关系, 分别做两种操作: A1. 三角乘法更新:(出边模式) 将 分别通过不同线性层投影,并用 gating (一般是元素乘法),后对所有k的结果求和,最后线性投影并附加到原来的表征上,用数学语言可写为: 其中 是可学习的权重矩阵,σ 是sigmoid激活函数,⊙ 代表逐元素乘法 A2. 三角乘法更新(入边模式) 将A1的 换成
B. 三角自注意力
- 关注起点
此模式更新边 Zij 时,将
i视为“起始节点”或锚点,并让 Zij 去“关注”所有其他从i出发的边 Zik。
其计算流程遵循标准的注意力机制:
- 查询 (Query): 由 经过线性变换 得到:。
- 键 (Key): 由所有 经过线性变换 得到:。
- 值 (Value): 由所有 经过线性变换 得到:。
- 注意力权重: 注意,这里引入了来自Pair表征 的偏置项,进一步加强三角关系。
- 加权求和: 6. 最终更新: 关注终点的注意力,把 换为 即可
虽然前几层 EvoFormer 就已经足以给出一个初步的结构原型,但是需要十几层的EvoFormer 来针对复杂蛋白质做不断地优化,进而锁定一个全局最优解
需要注意的是,类似Transformer,这里每一步都会使用一个残差链接,进而避免模型在层数过大时性能下降
Structure Moddule
在 EvoFormer 模块处理信息后, 将 single 表征(MSA的第一行, 目标氨基酸序列表征)加上 Pair 表征输入 structure Module 预测蛋白质
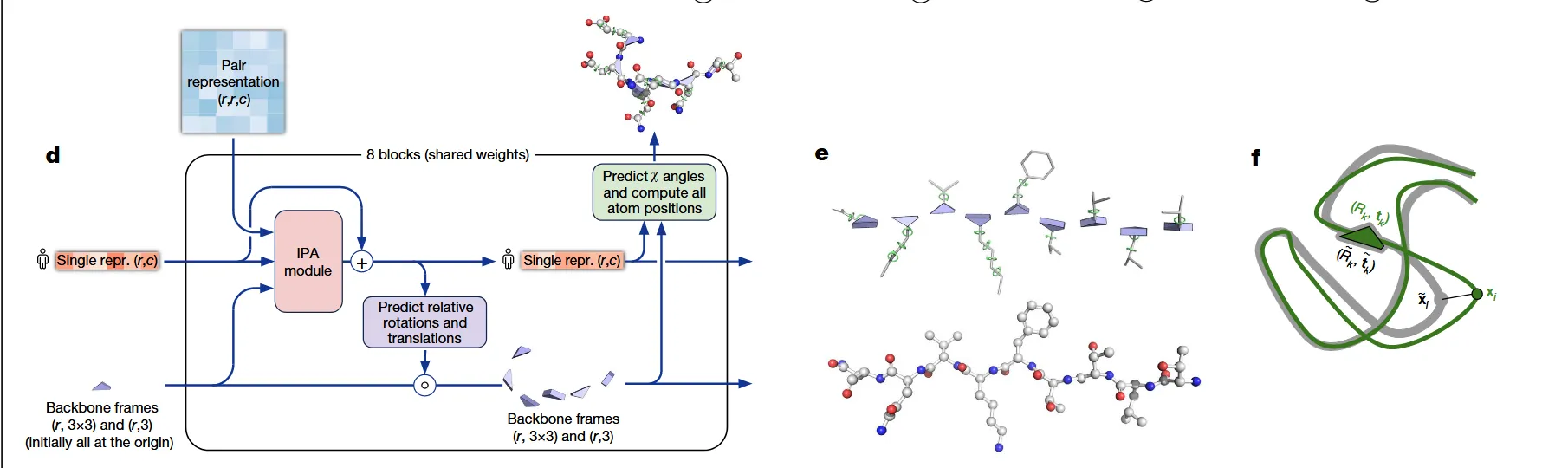
这里的一个核心想法是将 蛋白质视为 由 个独立 刚体 组成的 气体,而每一个残基 i 的骨架都被视为一个独立的局部坐标系(参考图 e),由一个旋转矩阵 表示朝向, 一个平移向量表示位置。
如果你有一个点在残基的局部坐标系中的坐标是 ,那么你可以用旋转矩阵和平移向量把它变换到世界坐标系中:
预测三维结构的核心模块是 IPA,这是是一种为三维几何空间“量身定做”的注意力机制。它的目标是让每个残基“环顾四周”,观察其他残基在3D空间中的位置,然后根据这些观察来更新自身的特征信息(即Single表征)。
以下是 AI提取、整理的关于 IPA以及后续预测、训练操作的细节:
第1步:在“局部”生成查询、键、值点
- 对于每一个残基
i,网络会利用其当前的Single表征(一个1D特征向量),通过几个不同的线性层,生成一组3D点。这些点是该残基的查询点(Query points)、键点(Key points)和值点(Value points)。 - 关键:这些点的位置坐标是在残基
i自身的局部坐标系中定义的。想象每个乐高积木表面都有几个固定的“触点”,这些触点的位置是相对于积木自身的。
第2步:将所有点“投影”到同一个世界
- 现在,模型使用每个残基
i的框架 ,将它所有的局部点(查询、键、值点)全部变换到同一个全局世界坐标系中。 - 这样一来,原先分散在各自局部坐标系中的点,现在都“漂浮”在同一个三维空间里,可以直接比较它们之间的距离了。
第3步:在“世界”中计算注意力
- 这是IPA与标准注意力的核心区别。它不使用向量点积来计算相似度,而是使用点与点之间的空间距离。
- 查询点(来自残基
i)和键点(来自残基j)之间的注意力分数,是基于它们在全局坐标系中欧氏距离的平方计算的。 - 不变性来源:因为三维空间中两点间的距离是一个几何不变量,无论你如何旋转或平移整个蛋白质(即改变所有点的全局坐标),它们之间的相对距离永远不变。因此,基于距离计算出的注意力分数也是不变的,这就是IPA“不变性”的来源。
- 计算出的注意力权重,被用来对**全局的值点(Value points)**进行加权求和,得到一个聚合后的几何信息。
第4步:将信息“收回”到局部
- 上一步得到的聚合信息是在全局坐标系中的。现在,模型需要将这个信息“分发”回每个残基。
- 对于每个残基
i,这个全局的聚合信息会被逆向变换,放回到它自身的局部坐标系中。
第5步:更新Single表征
- 最后,这个被收回来的、蕴含了丰富3D环境信息的局部几何信息,会与残基
i原始的Single表征融合(通常是拼接后通过一个前馈网络),生成一个更新后的、信息更丰富的Single表seminar。
C. 等变结构更新 (Equivariant Structure Update)
等变性 (Equivariance): 在IPA更新了Single表征之后,模型会根据这个新的Single表征来计算对每个残基框架 的更新量。这个更新是在每个残基的局部坐标系中计算和应用的,这使得整个结构更新操作对于全局的旋转和平移是等变的 。这意味着,如果将输入的蛋白质整体旋转,输出的更新后的蛋白质也会相应地整体旋转,而其内部结构不变。
D. 最终输出
经过8个迭代模块的更新后,网络利用最终的Single表征预测出每个残基的侧链扭转角 。结合最终的骨架框架 和这些侧链角度,就可以构建出蛋白质完整的、包含所有原子的三维结构。
训练策略
- 循环 (Recycling):迭代求精法 。模型的一次完整预测输出(MSA表征和Pair表征)不会被直接丢弃,而是被重新送回网络的第一层,作为下一次预测的额外输入。这个过程会重复3次,极大地提升了准确性
- 自蒸馏 (Self-Distillation):这是AlphaFold 2利用海量未标记序列数据的关键。
- 首先,用一个在PDB上训练好的模型,对来自Uniclust30数据库的大约35万个蛋白序列进行结构预测 。
- 然后,筛选出其中高置信度的预测结果,形成一个庞大的、由AI生成的结构数据集。
- 最后,从头开始训练一个新模型,其训练数据由75%的AI生成结构和25%的PDB真实结构混合而成 。 这个过程极大地提升了模型的准确性和泛化能力。